Lysosomal Disorders
Lysosomal disorders (LDs) are a group of over 70 inherited diseases caused by dysfunction of lysosomal proteins. Although every individual LD is a rare disorder, all LDs have a combined prevalence of 1 in 5.000 live births making them the most common cause of neurodegeneration in childhood and infancy. The major hallmark of the underlying lysosomal pathology is accumulation of macromolecules resulting in lysosomal hypertrophy, reduced degradative activity, impairment of autophagy and vesicular trafficking thereby affecting signaling pathways and cellular function. LDs are progressive, systemic disorders with a variable degree of neurological impairment. In childhood, neurological symptoms comprise developmental delay, seizures, movement disorders and regression. Furthermore, lysosomal dysfunction plays a role in neurodegenerative disorders in adulthood with underlying common features in cellular pathophysiology. Details on disease mechanisms, presentation, biomarkers and trajectories of disease progression are mostly incomplete. Only few therapies are available for LDs, mostly partially effective. All require early initialization to decelerate disease progression.
Overview
Phenotypic description of the first of lysosomal diseases (LDs) dates back to the the 19th century, before the lysosome was initially discovered as degradative organelle in 1955 and 1956 by de Duve et al. and Novikoff et al. Starting from the 1960s awareness arose that LDs are caused by enzymatic deficiencies leading to storage material accumulation. Since then, knowledge about lysosomal biology and LD pathophysiology is constantly growing and ultimately led to therapies for LDs. To date, enzyme replacement therapies, hematopoietic stem cell transplantation, substrate reduction therapies and pharmacological chaperones are licensed LD therapies. They mostly prevent disease progression but are hardly able to reverse signs and symptoms in patients. Gene therapies are on the verge of clinical application with one recently licensed for the treatment of Metachromatic Leukodystrophy. Despite growing efforts, understanding of the LD pathophysiology remains incomplete and a high unmet need for therapies for LDs persists.
Research Focus
Intracelluar disease pathology and therapy development
We aim for developing therapies for LDs. Elucidation of the underlying pathophysiology of LDs allows the identification of targets for therapeutic intervention. Untargeted screening for therapeutic agents in LDs followed by working up the detailed intracellular mode-of-action complements the search for therapies and generates insight into disease pathology vice versa. We make use of LD disease models in cell lines and animal models (mice, zebrafish) and patient derived fibroblasts and iPSCs for in-vitro and in-vivo research approaches. Experimental procedures comprise CRISPR/Cas9 gene editing, confocal and high-resolution microscopy, and high-throughput screening among others. Beside working on different LDs a main research focus are similarities of disease pathology across different LDs. We work closely with patient organizations and national- and international collaborators.
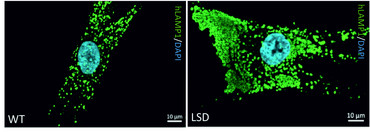
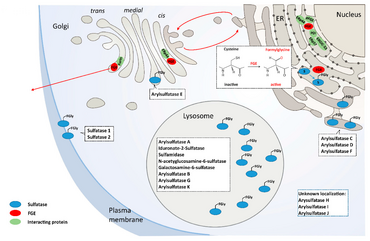
Our findings on Multiple Sulfatase Deficiency (MSD) highlight the translational research approaches towards a therapy for LDs in our group. MSD is an ultra-rare LD and is caused by mutations in the SUMF1 gene encoding the formylglycine generating enzyme (FGE). FGE is indispensable for full catalytic activity of all cellular sulfatases. Mutations in SUMF1 result in instable and functional compromised FGE leading to accumulation of storage material in lysosomal structures resulting in a phenotype comprising alterations in the autophagic flux and in secondary and tertiary storage phenomena.
MSD patients display a complex neurologic phenotype with high burden of systemic disease comprising combined clinical signs of single sulfatase deficiencies. No disease modifying treatments (DMT) exists for MSD despite high unmet need for MSD patients. Applying a high throughput screening for approved drugs we identified 3rd generation retinoids tazarotene and bexarotene to reverse MSD disease pathology in vitro. While we are elucidating the intracellular mode-of-action to identify alternative targets for drug treatment we are currently working towards a phase I/II clinical study for tazarotene’s use in MSD patients because both drugs are amenable to drug repurposing.
Publications
Cooperations:
Children’s Hospital of Philadelphia, USA, Rebecca Ahrens-Nicklas, Laura Adang, Xinying Hong
University of Cambridge, UK, Angeleen Fleming, David Rubinsztein
University of Southampton, UK, Matthias Baud
University of Bielefeld, Germany, Karthikeyan Radhakrishnan, Hartmut Niemann
Fraunhofer ITMP, Germany, Stefan Jacobs, Christian Brueser, Ole Pless, Aimo Kannt, Tanja Rossmanith
University of Washington, Seattle, USA, Michael Gelb, Zack Herbst
REMEDI4ALL
Funding
EU Horizon Europe research and innovation programme
Health Research Charity Initiatives/ Health Research Board Ireland (HRBI/HRC)
MSD Action Foundation, Irleland
CuraMSD, Spain
United MSD Foundation, USA
Una Cura Para Alma, Argentinien
Research Team
Principal Investigators
PD Dr. med. Lars Schlotawa

Lars Schlotawa graduated in medicine and received his MD degree from the University Medical Center Goettingen (UMG) for his work on GnRH receptor initiated signal transduction in endometrial cancer cell lines in 2005. During his internship in paediatrics and neuropaediatrics at UMG’s department of paediatrics and adolescent medicine he started his work on MSD through a collaboration with the UMG Institute of Biochemistry. In 2014 he started a post doc position at the University of Cambridge, UK, at the Cambridge Institute for Medical Research. Since 2018 he is working as a consultant for paediatrics, neuropaediatrics and metabolic medicine and as translational researcher and PI at UMG’s department of paediatrics and adolescent medicine. Lars received his venia legend in paediatrics in 2021 for his work on MSD.
contact: lars.schlotawa(at)med.uni-goettingen.de
Postdoctoral Fellows
Dr. rer. nat. Karolina Matysiak
Karolina completed her master’s degree in Biotechnology at the Jagiellonian University in Krakau, Polen. During her master thesis, she focused on a novel anti-inflammatory protein MCPIP1 (monocyte-chemotactic protein – induced protein 1) and created an animal model with a specific overexpression of MCPIP1 protein in adipose tissue. In 2014, she started her doctoral studies at the Hannover Medical School, at the Department of Clinical Biochemistry, where she was able to deepen her studies on MCPIP1 in context of Diabetes. Since 2019, she is working as a post doc at University Medical Center Goettingen. Her main research interest is the development of therapeutic approaches for very rare lysosomal storage disorders, with the focus on MSD.
contact: karolina.tyka(at)med.uni-goettingen.de
PhD/MD students/TAs
PhD Students:
Lina Schmidt
MD Students:
Marten Bertram
Lilly Hutzschenreuter
Lea Keufen
Technical Assistants:
Kathrin Schreiber
Tanja Wilke