Neurometabolic and Neurodegenerative Diseases
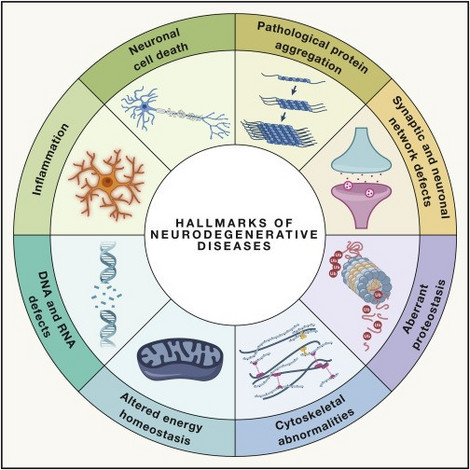
Pediatric neurodegenerative and neurometabolic diseases encompass a diverse group of rare disorders, with over 200 known variations so far. Affected children typically show normal development initially, but depending on the form of disease, they progressively lose motor, cognitive, auditory, and visual skills, often accompanied by seizures. These diseases often result from genetic disorders impacting brain metabolism, leading to the absence of essential substances or toxic waste accumulation, resulting in neuronal death. Many childhood neurodegenerative diseases are incurable, and unfortunately, over a third of affected children lack a known genetic cause.
Figure: Hallmarks of neurodegenerative diseases (Wilson et al., 2023)
Overview
The field of pediatric neurodegenerative diseases is extensive, encompassing more than 200 recognized forms with various clinical presentations as well as different genetic cause. These disorders include for instance aminoacidopathies, creatine disorders, mitochondrial cytopathies, peroxisomal disorders, and lysosomal storage disorders. Some forms are so rare that only a handful of cases have been reported worldwide, necessitating collaborative efforts for comprehensive research and data sharing. The genetic causes of many of these diseases are still unclear. Affected children and adolescents typically exhibit normal development initially but eventually experience a progressive decline in motor and cognitive skills, along with hearing and visual impairments, often accompanied by seizures. This deterioration results in disruptions or complete loss of these abilities, highlighting the challenging nature of these diseases. Many affected children experience genetic abnormalities that disrupt brain metabolism, resulting in the absence of crucial substances necessary for the structure, maintenance, and functionality of brain tissue and neurons. Alternatively, toxic metabolites may accumulate in the brain due to impaired breakdown processes. Consequently, these malfunctions lead to neuronal damage and eventual cell death. While there are effective therapeutic approaches available for some childhood neurodegenerative diseases, the majority of these conditions remain incurable at present. The identification of the genetic cause of the disease and subsequent functional analyses in different model systems enable us to gain valuable insights regarding general and basic mechanisms in neurometabolism and -inflammation. Thus, these results are potentially transferable to other pediatric neurodegenerative diseases and might reveal novel biomarkers. Understanding of what happens at disease onset and identification of possible prerequisites or external triggers is fundamental to develop the right surveillance and treatment strategies for our individual patients. Our aims are the improvement of diagnosis, implementation of suitable monitoring methods, better prediction of disease course and development of targeted new treatments for affected children.
Research Focus
Identification of New Disease Genes in Unclear Neurodegenerative Diseases
Identifying the genetic cause of neurodegeneration in children is crucial for effective therapy, prognosis, and genetic counseling, yet it remains elusive for many rare neurodegenerative disorders. This research focuses on utilizing advanced techniques such as Next-Generation Sequencing (NGS) to unravel the genetic basis of disease. Currently, Trio-whole exome sequencing, involving affected children and their parents, represents the state-of-the-art approach, while genome data and RNA-Seq are also being utilized. A team of skilled physicians and biologists analyze the generated data in conjunction with clinical information. Filtering strategies prioritize rare pathogenic de novo mutations, mutations affecting protein structure or splice sites.
The research aims to discover new disease-associated genes, expand the mutation spectrum of known disease-associated genes, and establish novel genotype-phenotype associations. Experimental projects are designed to validate findings and characterizing the detected variants. Ultimately, this research project contributes to understanding complex neurodegenerative conditions by investigating the underlying genetic defects and their impact on brain metabolism. Through comprehensive genetic analysis and in-depth examination of affected individuals, novel insights into the pathogenesis of these diseases are sought, providing the basis for the development of new therapy options.
Publications
Wong K., et al. (2022). Mutations in TAF8 cause a neurodegenerative disorder. Brain.
Cooperations
Prof. Dr. Peter Nürnberg, Cologne Center for Genomics (CCG)
Dr. Holger Thiele, Cologne Center for Genomics (CCG)
Contact
Dr. rer . nat. Susann Diegmann
IMDDHH: Enhancing the Clinical Picture and Therapeutic Strategies
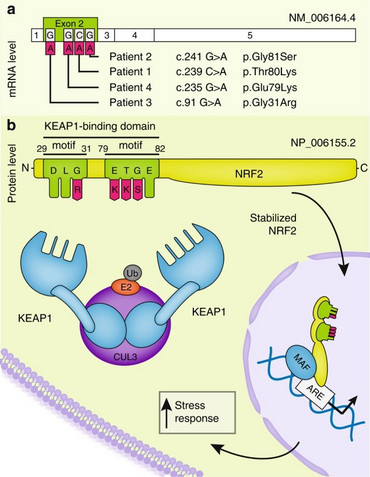
Our research group was the first to report the identification of de novo inborn mutations in NFE2L2, encoding the transcription factor NRF2, in four independent patients presenting with neurodegeneration. While somatic mutations in NFE2L2 have been associated with promoting cell survival and drug resistance in cancer, we have discovered that these same mutations, occurring as inborn de novo mutations, lead to an early onset multisystem disorder characterized by failure to thrive, immunodeficiency, and neurological symptoms (OMIM: IMDDHH #617744). The pathological NRF2 accumulation results in widespread dysregulation of gene expression and disruption of the cytosolic redox balance.
To comprehensively understand the disease and enhance our clinical understanding, expanding the NRF2-affected patient cohort is essential for gaining insights into the mutation spectrum. Furthermore, our research investigates the therapeutic potential of NRF2 inhibitors, commonly used in cancer treatment, as viable options for this disorder. Additionally, our ongoing investigation focuses on identifying robust biomarkers for rapid and efficient diagnosis. Through this research, we highlight the importance of NRF2 mutations in driving a multisystem dysfunction, expanding our understanding of its role beyond cancer and opening avenues for early intervention and treatment strategies.
Publications
Cooperations
Prof. Dr. med. Peter Huppke, Jena
Prof. Dr. Peter Nürnberg, Cologne Center for Genomics (CCG)
Dr. Holger Thiele, Cologne Center for Genomics (CCG)
Contact
Dr. rer. nat. Susann Diegmann
Neuronal Ceroid Lipofuscinosis (NCL)
Neurometabolic and neurodegenerative diseases of childhood are very rare and mostly caused by single, recessive inherited mutations of functionally important proteins. Although many of the related disease-causing mutations have been known for a long time, in many cases little is known about the underlying molecular pathomechanisms. A detailed understanding of the molecular biological and biochemical processes involved in the onset and progression of these diseases provides an important basis for the development of therapeutic approaches. In cooperation with other research groups, we have elucidated the protein structures of several disease related lysosomal enzymes and investigated the structural-biological and functional consequences of known mutations. Based on our structural data we developed hypotheses for enzymatic and functional mechanisms and refined these concepts experimentally.
Another research project concerns the quantifiable laboratory aspects of neurometabolic diseases. Biomarkers play a crucial role in the diagnosis and disease progression monitoring of neurometabolic and neurodegenerative disorders. To identify novel biomarkers, we apply both classical laboratory diagnostic methods as well as untargeted/targeted metabolomics.
Batten disease, also known as neuronal ceroid lipofuscinosis (NCL), is a rare and devastating neurodegenerative disorder that primarily affects children. It is characterized by the accumulation of lipopigments (lipofuscin) in the brain and other tissues and progresses with loss of vision, seizures, cognitive decline, and motor impairment. The disease is caused by genetic mutations that affect the function of lysosomes, cellular compartments responsible for cellular waste removal and recycling processes. Currently there is no cure for Batten disease, and treatment mainly focuses on managing symptoms and providing supportive care.
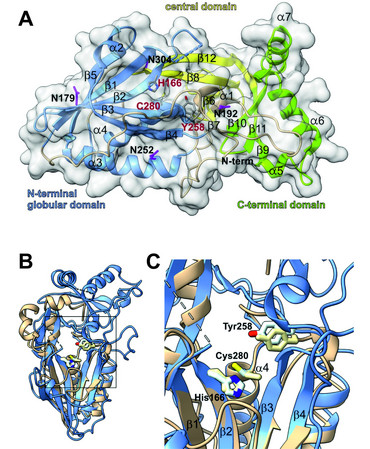
NCL5, the finnish late infantile variant of Batten disease, is triggered by mutations in the CLN5 gene. While the first disease causing mutations in human related to NCL5 were published around 1998, the structure and function of the CLN5 protein remained unclear until recently. In cooperation with other groups from the University of Göttingen and Max Planck Institute for Multidisciplinary Sciences we were able to provide the crystal structure of CLN5. We further showed that CLN5 acts as a depalmitoylase, cleaving thioester bonds between SH-groups of surface located cysteine residues conjugated to palmitate. Our current research focuses on the identification of the natural CLN5 substrates and the pathomechanistic role of CLN5 in the onset and progression of Batten disease.
Publications
Cooperations
Prof. G.M. Sheldrick, University of Göttingen
Dr. Steffen Becker, Max Planck Institute for Multidisciplinary Sciences
Dr. Christof Lenz, UMG
Contact
Priv.-Doz. Dr. rer. nat. Ralph Krätzner
Oxidative and Mitochondrial Stress in Neurometabolic Diseases
The connection between oxidative stress and mitochondrial dysfunction in neuroinflammatory and degenerative diseases as well as in inborn errors of metabolism (IEM) is well established in literature. Large parts, however, still remain unknown regarding their causality, direct impact on pathophysiology of disease onset, progression and repair. Using the example of individual specific diseases, such as X-linked adrenoleukodystrophy, we investigate factors causing oxidative stress in health and disease as well as potential protective agents. Mitochondrial stress and dysfunction are found in many not primarily mitochondrial diseases. Through analysis of mitochondrial function and structure in neurometabolic diseases we aim to elucidate their distinct role in pathophysiology and improve understanding of interplay between cellular organelles in healthy and disturbed state. Understanding these pathophysiological mechanisms in detail will help to design new therapeutic strategies and to develop new treatments by targeting specific neurometabolic pathways.
Cooperations
Malte Tiburcy, Department of Pharmacology and Toxicology, UMG
Branimir Berečić, Department of Pharmacology and Toxicology, UMG
Christian Brüser, Fraunhofer Institute for Translational Medicine and Pharmacology
Jan Keller-Findeisen, Fraunhofer Institute for Translational Medicine and Pharmacology
Contact
Dr. med. Lara Marten
Lipidomic Profiling in Early Onset Myelin Disorders
While the understanding of lipid metabolism and the role of complex lipids in neurometabolism and -inflammation is increasing, the extent of unknown aspects and connections remains large. The brain, especially regions with high myelin content, is a lipid rich organ. Also, lipids are substantial for inflammatory processes throughout the body in physiological as well as in pathophysiological conditions. Dysregulation of lipid metabolism, as for example caused by genetic conditions affecting lipid synthesis or metabolization, can have tremendous impact on inflammatory reactions. On the other hand, damage to lipid containing tissues, for example through demyelinating processes, will lead to lipid containing debris, which is potentially detectable in tissue and body fluids. We investigate the role of lipid metabolism in early onset myelin disorders and implications for neuroinflammation and degeneration through lipidomic analysis, which is a promising tool and applicable to different tissues and body fluids.
Cooperations
Stefan Nessler, Institute of Neuropathology
Christine Stadelmann, Institute of Neuropathology
Dirk Fitzner, Department of Neurology
Justus Lattau, Department of Neurology
Funding
TRR274 – Advanced clinician scientist module
Contact
Dr. med. Lara Marten
Research Team

Principal Investigators
Prof. Dr. med. Jutta Gärtner

Director, Department Pediatrics and Adolescent Medicine
Contact: kinderklinik(at)med.uni-goettingen.de
Priv.-Doz. Dr. rer. nat. Susann Diegmann

Susann Diegmann, a scientist specializing in the genetic complexities of human diseases. She studied Biotechnology and completed her PhD at the Institute of Pathology in Würzburg, Germany, supervised by Prof. Andreas Rosenwald. Her research focused on Multiple Myeloma's complex genetic network. Since beginning her postdoc in the lab of Prof. Jutta Gärtner at UMG in 2015, Susann has been identifying genes related to neurometabolic and neurodegenerative disorders in children and adolescents. Skilled in Next Generation Sequencing (NGS), she collaborates with pediatricians to identify pathogenic variants. Susann continues to characterize these genes, designing functional projects to apply her findings. An invaluable team member, she's advancing our understanding and treatment of these conditions.
contact: susann.diegmann(at)med.uni-goettingen.de
Priv.-Doz. Dr. rer. nat Ralph Krätzner

Ralph Krätzner obtained his PhD in the group of Prof. Sheldrick, chair for structural chemistry, Inst. of Inorganic Chemistry, University of Göttingen. His thesis was about structural investigation of variants of the Ecballium elaterium trypsin inhibitor-II (EETI-II). After post doc years in the department of Molecular Pharmacology, University Medicine Göttingen (UMG), he joined the Clinic of Pediatrics and Adolescent Medicine as head of the lab facilities. Ralph Krätzner obtained the “venia legendi” from UMG, he was recognized as European specialist in laboratory medicine (EuSpLM) by the EFLM in 2019.
contact: ralph.kraetzner(at)med.uni-goettingen.de
Dr. med. Lara Marten

Lara Marten obtained her Dr. med. from the University of Hamburg with a project in the Laboratory of Cellular and Molecular Radiation Oncology at Harvard Medical School/Massachusetts General Hospital (Boston, USA) supervised by Henning Willers and Jochen Dahm-Daphi. Her project focused on the impact of tumor suppressor p53 on DNA repair mechanisms. After finishing pediatric residency at the University Medical Center Hamburg-Eppendorf, she concluded a research fellowship at the Department of Physiological Chemistry, University of Veterinary Medicine Hannover with the project “Characterization of new mutant variants of intestinal lactase-phlorizin hydrolase”, supervised by René Santer and Hassan Naim. Since May 2020 Lara Marten is part of the UMG team and participated in the advanced clinician scientist program of TRR 274 “Checkpoints of Central Nervous System Recovery” with the project “Role of neurometabolism in demyelination and repair processes in early onset myelin disorders”, funded by DFG.
contact: lara.marten(at)med.uni-goettingen.de
Dr. med. Kolja Meier
Kolja Meier analyzed network activity in the mouse hippocampus during his PhD in medicine at the Center for Molecular Neurobiology in Hamburg in the research group Experimental Neuropediatrics of Prof. Dirk Isbrandt. After his PhD he began his residency in pediatrics in Göttingen. As early clinician scientist in the TRR274 he develops postprocessing pipelines for multimodal MRI data of patients with neuroinflammatory and neurometabolic diseases, such as pediatric MS and X-linked adrenoleukodystrophy. Furthermore, he is further engaged in the analysis of trio-exome data to solve cases of patients with unclear neurometabolic diseases.
contact: kolja.meier(at)med.uni-goettingen.de
Prof. Dr. med. Lars Schlotawa

Lars Schlotawa graduated in medicine and received his MD degree from the University Medical Center Goettingen (UMG) for his work on GnRH receptor initiated signal transduction in endometrial cancer cell lines in 2005. During his internship in paediatrics and neuropaediatrics at UMG’s department of paediatrics and adolescent medicine he started his work on MSD through a collaboration with the UMG Institute of Biochemistry. In 2014 he started a post doc position at the University of Cambridge, UK, at the Cambridge Institute for Medical Research. Since 2018 he is working as a consultant for paediatrics, neuropaediatrics and metabolic medicine and as translational researcher and PI at UMG’s department of paediatrics and adolescent medicine. Lars received his venia legend in paediatrics in 2021 for his work on MSD.
contact: lars.schlotawa(at)med.uni-goettingen.de
Dr. med. Matthias Kettwig

Matthias Kettwig obtained his PhD in medicine in the lab of Prof. Rehling at the Institute for Biochemistry, Göttingen. His doctoral work focused on the functional analysis of a new identified lysosomal matrix protein. He participate in the clinician scientist program by Göttinger Kollegs für Translationale Medizin der Universitätsmedizin Göttingen, gefördert vom Niedersächsischen Ministerium für Wissenschaft und Kultur, http://www.kolleg-transmed.med.uni-goettingen.de/
Matthias investigates the pathophysiological mechanism of interferon-driven neuroinflammation in RNASET2-deficient mice.
contact: matthias.kettwig(at)med.uni-goettingen.de
PhD/MD students/ Technical Assistance
PhD Student: Ina Götz
MD Student: Maxi Lüttgens
Technical Assistance: Corinna Dickel, Ellen Krämer