Type I Interferonopathies
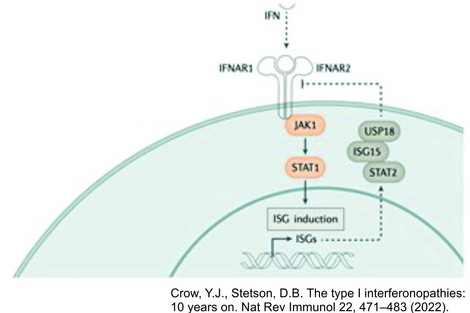
As one of the most relevant cytokines in the innate immune response, type I interferon plays a central role in the pathophysiology of inflammation-related CNS damage. Understanding this fundamental role of type I interferon upregulation as a common feature in a set of Mendelian inborn errors of immunity leads to the definition of type I interferonopathies as an independent disease group in 2011. Over the last 10 years more than 40 discrete genotypes were defined as type I interferonopathies with a broad associated phenotypic spectrum.
You can find a request form for the diagnostic evaluation of interferonopathies (interferon score) available for download here.
Ein Anforderungsformular zur Diagnostik von Interferonopathien (Interferon-Score) zum Download finden Sie hier.
Overview
Type I interferon is an important cytokine of protective immune response in humans in general. As a key element of the innate immune system, cytokines produced by infected or damaged cells are of great importance especially in the CNS. Thereby interferon is the main mediator of antiviral immune response and so central to protect the CNS from viral infections.
The harmful effect of interferon in humans is already known since more than 40 years. In the late 90s the first genetic disorder with increased levels of interferon activity in the serum and cerebrospinal fluid of children that resembled an in utero-acquired viral infection was found and now referred to as Aicardi–Goutières syndrome (AGS).
In 2011 the rising understanding of the immune system and the type I interferon signaling in human disease leads to the coining of the term type I interferonopathy as it serve as a defining feature of a novel set of Mendelian inborn errors of immunity.
In the last 10 years the number of type I interferonopathies increase in up to 40 discrete genotypes by expert clinical phenotyping, the use of screening assays and the advent of next-generation sequencing.
This rapid increase in knowledge has both informed and been informed by fundamental discoveries in innate immunity, leading to a better understanding of the role of type I interferons in health and disease, and the possibility of therapeutic initiatives aimed at limiting type I interferon signalling.
Research Focus
Interferon-driven neuroinflammation
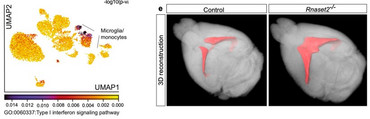
As the first described whole-body mouse model of type I interferonopathy with a CNS phenotype, the Rnaset2−/− mouse provides important insights into the mechanisms of interferon-I- dependent neurodegeneration.
Rnaset2−/− mice develop a severe type I interferon-driven neuroinflammatory disorder, which is characterized by elevated interferon-stimulated genes (ISGs), perivascular infiltrates of inflammatory monocytes, activated microglia, and diffuse infiltration of CD8+ T cells of the effector memory type throughout the entire CNS. The perivascular inflammation leads to blood-brain barrier leakage as shown by gadolinium enhancement on cerebral magnetic resonance imaging (MRI). Furthermore, the observed neuroinflammation might foster the documented general brain atrophy, which is accentuated in the hippocampus. Not surprisingly, memory function is impaired in these mice.
As shown by single nuclei RNA sequencing, type I interferon signaling in the CNS, is accompanied by a homeostatic dysfunction of glial cells (microglia, astrocytes, oligodendrocytes, and OPCs) and neurons. In order to improve the mechanistic understanding of type I interferon-driven neuropathological changes, we need to better understand how the observed transcriptional disturbances in glial cells and in neuronal subclusters contribute to the disease pathogenesis.
Publications
Cooperations
Dr. Stefan Nessler, Institute of Neuropathology
Prof. Dr. Oliver Wirths, Clinic for Psychiatry and Psychotherapy
RNASET2-deficient cystic leukoencephalopathy
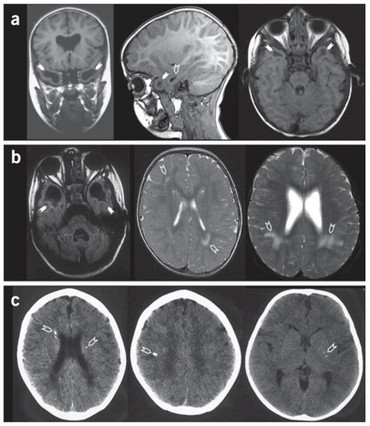
Human RNaseT2 deficiency is a rare, monogenetic, early childhood onset cystic leukoencephalopathy (CLE) which manifests as psychomotor delay, spasticity, epilepsy, and normo- or microcephaly during the first year of life. Brain magnetic resonance imaging (MRI) reveals frontal and temporal lobe cystic lesions, multifocal white matter alterations, and cerebral atrophy. The phenotypic features of RNaseT2-deficient CLE are indistinguishable from the sequelae of in utero cytomegalovirus (CMV) brain infection. Moreover, there is a significant clinical and neuroradiological overlap of RNaseT2-deficient CLE with Aicardi-Goutières syndrome (AGS), with some affected individuals demonstrating cerebrospinal fluid (CSF) pleocytosis, elevated levels of CSF neopterin as an inflammatory marker, and an overexpression of interferon-stimulated genes (ISGs) in peripheral blood.
We are currently collecting more details on the clinical picture of RNaseT2-deficient CLE by evaluating clinical questionnaires, describing autoimmune phenomena and assessing detailed neuro images.
Interest in joining this collaborative project? Do not hesitate to contact us!
Publications
Research Team

Principal Investigators

Priv.-Doz. Dr. med. Matthias Kettwig

Matthias Kettwig obtained his PhD in medicine in the lab of Prof. Rehling at the Institute for Biochemistry, Göttingen. His doctoral work focused on the functional analysis of a new identified lysosomal matrix protein. He participate in the clinician scientist program by Göttinger Kollegs für Translationale Medizin der Universitätsmedizin Göttingen, gefördert vom Niedersächsischen Ministerium für Wissenschaft und Kultur, http://www.kolleg-transmed.med.uni-goettingen.de/
Matthias investigates the pathophysiological mechanism of interferon-driven neuroinflammation in RNASET2-deficient mice.
contact: matthias.kettwig(at)med.uni-goettingen.de
Postdoctoral Fellows
Dr. rer. nat. Katharina Ternka

Katharina completed her diploma in Biology at the University of Osnabrück and her PhD at the University of Düsseldorf. She deepened her knowledge in the neuroscience field as a postdoc in the research group of Prof. Roland Brandt at the University of Osnabrück. Since 2013 Katharina focused on the RNASET2-deficient cystic leucoencephalopathy at the University Medical Center Göttingen in the group of Prof. Jutta Gärtner. She generated a model for RNASET2-deficiency in mice and works passionately with this model to investigate the pathomechanisms of interferone-driven neuroinflammatory diseases.
Contact: katharina.ternka(at)med.uni-goettingen.de
Kristin Wendland, PhD

Kristin completed her bachelor's degree in Biosciences at the Universität Potsdam and her master's degree in Neurobiology and Behavior at the Freie Universität Berlin. She deepened her knowledge in the neuroscience field with a Phd in Medical Neurosciences at the Charité - Universitätsmedizin Berlin. Her main interest was the investigation of neuronal survival in the context of stroke. Thereby she gained extensive knowledge in wide-field and confocal microscopy through an Analytical & Quantitative Light Microscopy Course (AQLM) in Woods Hole Massachusetts, USA and a four month research stay at Sunnybrook Research Institute Toronto, Canada. In 2019, Kristin started her postdoc research in neuroinflammation at the Universitätsmedizin Göttingen with the ambition of elucidating the pathophysiological pathway of RNASET2-deficiency.
contact: kristin.wendland(at)med.uni-goettingen.de
PhD/MD students
Melissa Ballüer
Luise Block-Gruppe
Fynn Heinemann
Paul Hock
Milena Irsfeld
Greta Küppers
Leonie Kadel
Yannick Stinus
Alumni
Lucia Gest (Bachelor)
Pascal Götz (cand. med.)
Charlotte Schob (Bachelor)
Carlotta Seifert (cand. med.)